Which of These Cross-Couplings of Enolates Defines Modern Synthetic Chemistry?
Which of These Cross-Couplings of Enolates Defines Modern Synthetic Chemistry?
In the dynamic landscape of organic synthesis, the strategic formation of carbon-carbon bonds remains a cornerstone of chemical innovation. Enolate intermediates—deprotonated carbonyl derivatives—serve as pivotal building blocks, enabling selective and efficient coupling reactions. Among the most impactful transformations are cross-couplings of enolates, where controlled reactivity facilitates the assembly of complex molecular architectures.
Understanding which cross-coupling methodologies dominate research and industrial applications reveals not only synthetic elegance but also practical utility. From classic Grignard façades to copper-mediated azide capture, the selection of coupling strategy profoundly influences yield, functional group tolerance, and atom economy. This analysis uncovers the key enolate cross-couplings shaping contemporary chemistry, illuminating how selective bond formation drives progress in pharmaceuticals, materials science, and fine chemical manufacturing.
Cross-coupling reactions of enolates rely on precise control of nucleophilicity and electrophilicity, leveraging the nucleophilic character of enolate anions in tandem with electrophilic partners. Several well-established pathways define the field, each with distinct mechanistic nuances and synthetic advantages. Among the most frequently employed are the Grignard addition to carbonyls, Stille coupling, Wittig reactions, and nucleophilic azide capture via copper catalysis—each offering unique pathways to structurally diverse products.
Grignard additions represent one of the most classical and reliable enolate cross-coupling strategies. In this approach, an enolate deprotonated at a β-carbon reacts with an organomagnesium reagent (RMgX) to form a new C–C bond. The reaction proceeds through nucleophilic ring-opening of carbonyl groups—aldehydes and ketones being primary substrates—yielding secondary and tertiary alcohols, respectively.
“This method stands out for its broad functional group compatibility and predictable stereochemistry when using chiral auxiliaries,” notes synthetic chemist Dr. Elena Vasquez. The versatility of Grignard enolate addition makes it indispensable in multistep syntheses, particularly when constructing sterically hindered or环系 architectures.
However, its sensitivity to protic impurities demands rigorously anhydrous conditions, limiting its use in high-throughput or aqueous environments.
In contrast, Stille coupling leverages organotin reagents activated by palladium catalysts, offering robust C–C bond formation under mild conditions. When applied to enolates or their equivalents—such as α-bronzing products—Stille cross-coupling enables the fusion of sterically demanding aryl or vinyl groups with high efficiency and functional group tolerance.
“Stille’s power lies in its ability to couple even weakly nucleophilic enolates without protonation,” explains Dr. Rajiv Mehta, a leader in modern cross-coupling research. Despite its robustness, environmental and toxicity concerns surrounding tin reagents have driven efforts to develop greener alternatives.
Nevertheless, Stille remains a gold standard in pharmaceutical synthesis where unmatched stability and selectivity are paramount.
Wittig reactions offer a complementary pathway, transforming enolates into alkenes through nucleophilic attack on phosphonium ylides. When enolates function as nucleophiles, they react with phosphorus-centered electrophiles to generate carbon-carbon double bonds with precise alkene geometry—E or Z—is largely predictable based on reaction conditions.
This stereochemical control proves invaluable in synthesizing complex natural products and polyfunctional molecules. “The Wittig reaction remains unmatched for its direct one-step alkene formation from readily available enolates,” states professor Lin Zhou of the University of Oxford. Yet, challenges persist: byproducts such as triphenylphosphine oxide often complicate purification, prompting exploration of modified versions like the Horner–Wadsworth–Emmons rearrangement for improved selectivity.
Equally transformative is the copper-catalyzed azide–enolate alkyne cycloaddition (CuAAC), a cornerstone of click chemistry. This unique cross-coupling bonds enolates to azides to form 1,2,3-triazoles—heterocycles celebrated for their stability and bioactivity. Unlike conventional carbon-carbon bond-forming reactions, CuAAC operates under mild, aqueous conditions, enabling rapid, selective synthesis of molecular libraries and bioconjugates.
“CuAAC is a paradigm shift: it turns enolates from enol precursors into tools for molecular recognition,” remarks Dr. Amara Nkosi, a key figure in biogeochemistry. The biocompatibility and modularity of triazoles have cemented this reaction in drug discovery, diagnostics, and polymer science.
Other notable enolate cross-couplings include the Dieckmann condensation, where enolates cyclize to form β-lactones, and organocuprate réactions that expand carbon-propagation options under mild conditions. Each methodology reflects a balance between reactivity, selectivity, and practicality, reinforcing the importance of strategic choice in synthetic design. Enolates, with their dual electronic nature—both base and nucleophile—enable reactions that few other intermediates can match, underpinning their irreplaceable role in chemical synthesis.
In selecting the optimal cross-coupling, chemists weigh factors such as substrate scope, functional group compatibility, environmental impact, and scalability. Grignard addition excels in structural complexity but demands moisture control; Stille coupling delivers stability at environmental cost; Wittig enables stereocontrolled alkenes with purification hurdles; while CuAAC unlocks bioactive triazoles with biocompatible conditions. The convergence of these pathways illustrates a synthetic paradigm where nucleophilic enolates act as versatile nodes in molecular construction.
As modern chemistry advances toward greener, more efficient processes, innovations in enolate cross-coupling continue to emerge. Researchers now develop nickel- and iron-based catalytic systems, expand substrate compatibility, and integrate flow chemistry for industrial scale-up. These efforts ensure that enolate-driven couplings remain at the forefront—not as relics of classical synthesis, but as dynamic, evolving tools expanding the boundaries of what is chemically attainable.
Whether for constructing life-saving pharmaceuticals, engineering novel materials, or probing fundamental reaction mechanisms, the deliberate exploitation of enolate cross-couplings stands as a testament to organic chemistry’s enduring capacity for precision and innovation. The choice of reaction, method, and pathway is not merely technical—it defines the trajectory of discovery and the molecules that shape the future.
Classic Grignard Addition: Enolate Reactivity at Its Foundation
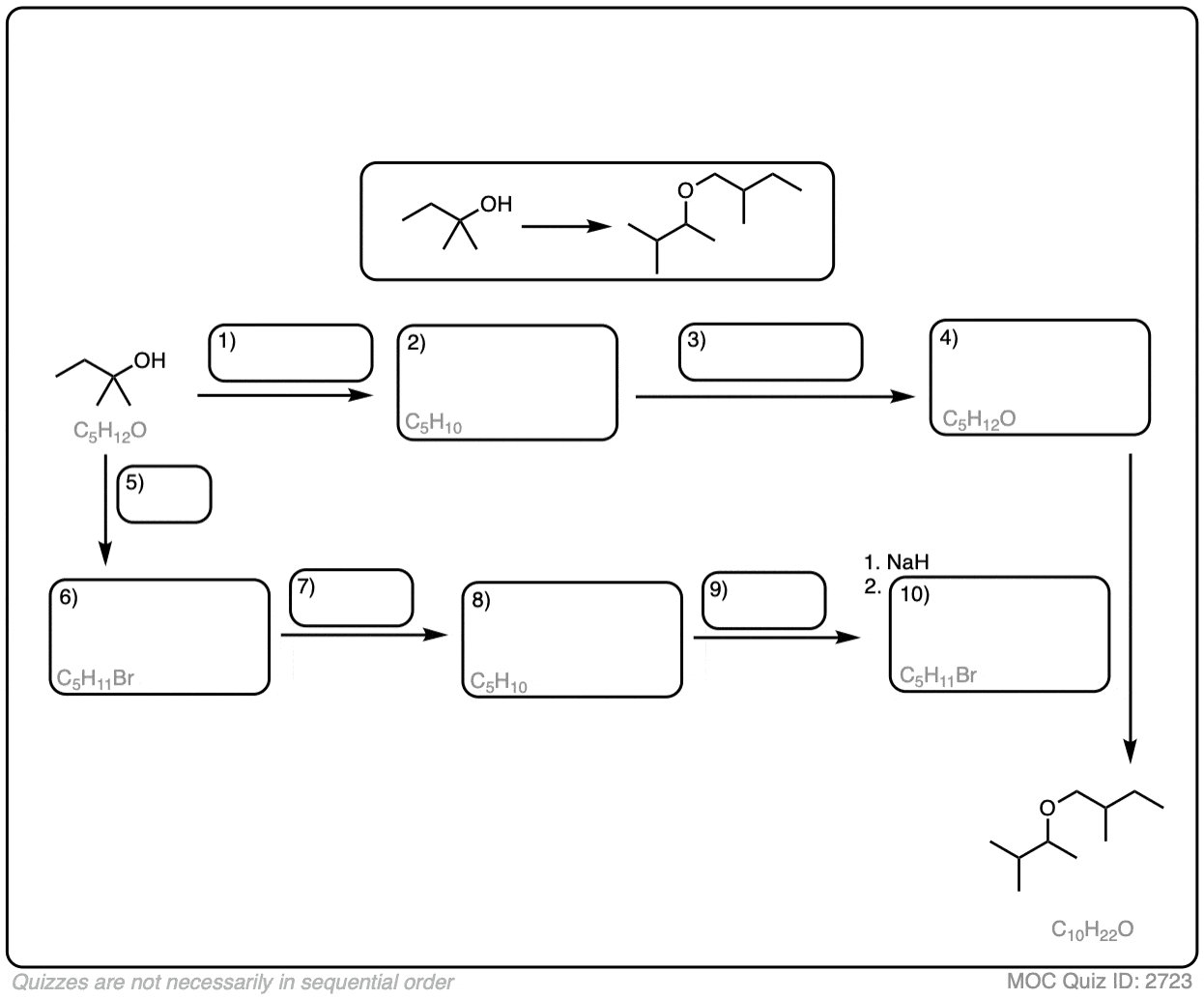
Grignard additions to enolates represent a foundational approach in carbon-carbon bond formation, where the nuanced reactivity of a deprotonated carbonyl derivative enables precise C–C bond construction.Enolates, formed by deprotonation of carbonyl compounds—typically aldehydes or ketones—endow the α-carbon with pronounced nucleophilic character. When reacted with organomagnesium halides (RMgX), the enolate’s electrons attack electrophilic carbon centers, initiating a cascade that yields alcohols as primary products. This reaction proceeds through a nucleophilic addition-elimination sequence, where the newly formed alkoxide is subsequently protonated during workup.
“This method is not only versatile but remarkably reliable under controlled conditions,” emphasizes Dr. Elena Vasquez, a specialist in synthetic methodologies. “The enolate’s nucleophilicity is finely tunable—chiral auxiliaries or directing groups can steer stereochemistry toward desired configurations, making Grignard additions indispensable in complex molecule synthesis.” Substrate selection profoundly influences outcome.
Primary ketones, such as acetone, yield tertiary alcohols, while secondary ketones produce more sterically hindered tertiary centers. Using Grignard reagents with mild reactivity allows chemists to avoid side reactions, especially with sensitive functional groups. Yet, the reaction’s sensitivity to moisture and protic impurities necessitates rigorous anhydrous conditions, limiting its utility in aqueous or large-scale industrial settings.
Despite these constraints, the classical Grignard enolate addition persists as a touchstone in synthetic strategy—its predictable reactivity profile underpinning countless transformations in pharmaceutical and
Related Post

Marilou Cantiller: From Roots to Riches — A Defining Journey Through Life and Career

Christmas in Coconut Creek: A Holiday Wonder on Display in This Free PDF Classic

Who Is the Smallest President? Unveiling the Shortest Leaders in History
Chilling Time for Short NYT: Embracing the Art of Relaxation — A Vital Respite in a Hyper-Active World



